Decoding DNA: A Comprehensive Guide to Sequence Encoding Techniques for Machine Learning Applications
Exploring Methods to Transform Genetic Sequences into Machine-Readable Formats
Decoding DNA: From Biological Sequences to Machine Learning Features
Visual representation of DNA sequence encoding methods for machine learning applications
Encoding DNA sequences into formats suitable for machine learning models is a critical step in genomic data analysis. The choice of encoding method can significantly impact model performance, computational efficiency, and biological interpretability. Various encoding techniques have been developed, each with its own strengths and weaknesses tailored to different types of genomic analyses.
🧬 Introduction
The transformation of biological sequences into numerical representations is fundamental to applying machine learning in genomics. DNA, composed of four nucleotides (A, T, G, C), presents unique challenges for computational analysis due to its discrete nature, variable length sequences, and complex biological relationships.
This comprehensive guide explores ten major encoding techniques, their applications, trade-offs, and implementation considerations for modern genomics research.
🔢 Classical Encoding Methods
1. One-Hot Encoding
Overview
The most fundamental approach where each nucleotide is represented as a binary vector. This method creates a sparse, high-dimensional representation that preserves exact sequence information.
Encoding Scheme: - A: [1, 0, 0, 0] - T: [0, 1, 0, 0]
- C: [0, 0, 1, 0] - G: [0, 0, 0, 1]
Strengths
- ✅ Complete Information Preservation - No loss of sequence data
- ✅ Simple Implementation - Straightforward and interpretable
- ✅ Universal Compatibility - Works with any ML algorithm
- ✅ Position Awareness - Maintains exact positional information
Weaknesses
- ❌ High Dimensionality - Creates very large matrices for long sequences
- ❌ Sparse Representation - Inefficient memory usage
- ❌ No Biological Context - Doesn’t capture nucleotide relationships
- ❌ Fixed Length Requirement - Sequences must be padded or truncated
Implementation Example
import numpy as np
from sklearn.preprocessing import LabelEncoder, OneHotEncoder
def one_hot_encode_dna(sequence):
"""One-hot encode DNA sequence"""
mapping = {'A': [1,0,0,0], 'T': [0,1,0,0],
'C': [0,0,1,0], 'G': [0,0,0,1]}
return np.array([mapping[nucleotide] for nucleotide in sequence])
# Example usage
sequence = "ATCG"
encoded = one_hot_encode_dna(sequence)
print(f"Sequence: {sequence}")
print(f"Encoded shape: {encoded.shape}")Best Use Cases
- Short sequences (< 1000 bp)
- Exact position matters (promoter analysis, binding sites)
- Interpretability required (regulatory element identification)
2. k-mer Tokenization
Overview
DNA sequences are segmented into overlapping or non-overlapping substrings of length ‘k’. This approach captures local sequence patterns and reduces computational complexity.
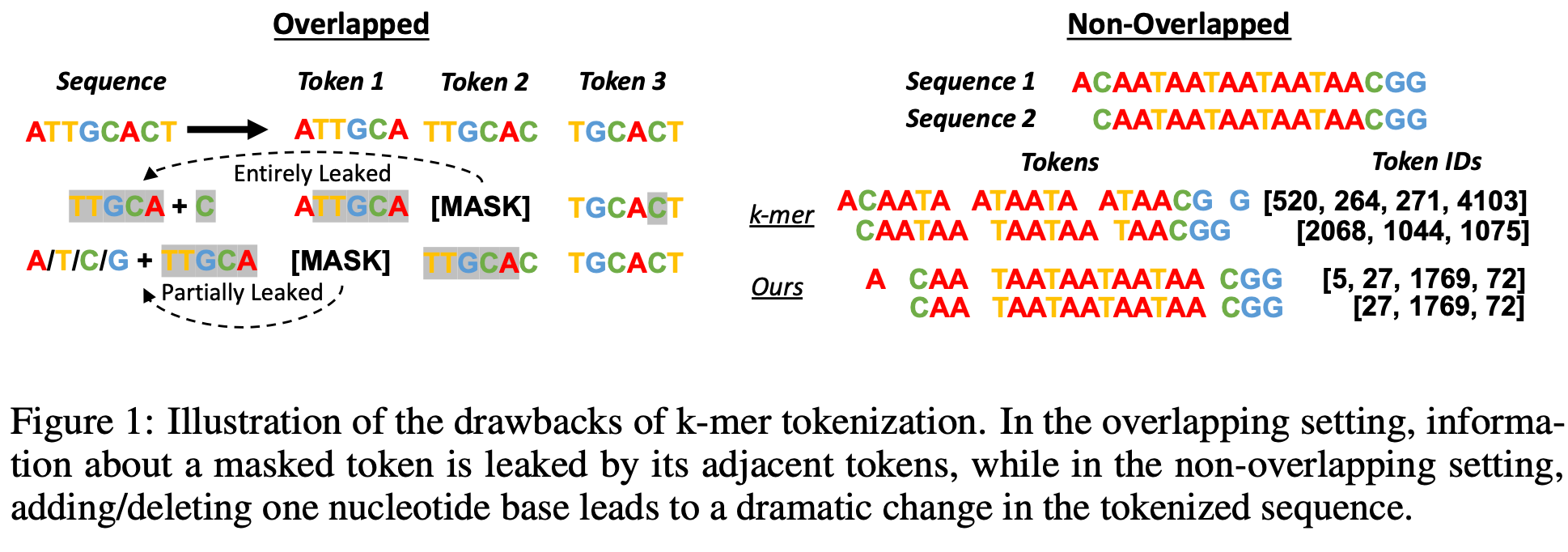
k-mer tokenization approach. Source: Zhou et al., DNABERT-2
Strengths
- ✅ Pattern Recognition - Captures local motifs and patterns
- ✅ Dimensionality Reduction - Reduces sequence length significantly
- ✅ Biological Relevance - k-mers correspond to biological motifs
- ✅ Flexible k-values - Adjustable for different applications
Weaknesses
- ❌ Information Leakage - Overlapping k-mers create redundancy
- ❌ Sample Inefficiency - Non-overlapping approach loses information
- ❌ Limited Context - May miss long-range dependencies
- ❌ k-value Selection - Requires optimization for each task
Implementation Example
def generate_kmers(sequence, k=3, overlap=True):
"""Generate k-mers from DNA sequence"""
if overlap:
step = 1
else:
step = k
kmers = []
for i in range(0, len(sequence) - k + 1, step):
kmers.append(sequence[i:i+k])
return kmers
# Example usage
sequence = "ATCGATCG"
kmers_3 = generate_kmers(sequence, k=3, overlap=True)
print(f"3-mers (overlapping): {kmers_3}")
kmers_3_no = generate_kmers(sequence, k=3, overlap=False)
print(f"3-mers (non-overlapping): {kmers_3_no}")Performance Considerations
- k=3: Good for local patterns, 64 possible tokens
- k=4: Balance of specificity and vocabulary size (256 tokens)
- k=5: High specificity, large vocabulary (1024 tokens)
- k=6: Very specific, may overfit (4096 tokens)
🤖 Advanced NLP-Inspired Methods
3. Byte-Pair Encoding (BPE)
Overview
An adaptive tokenization method that iteratively merges the most frequent character pairs, creating a vocabulary that balances granularity with efficiency. Originally from natural language processing, BPE has proven highly effective for genomic sequences.
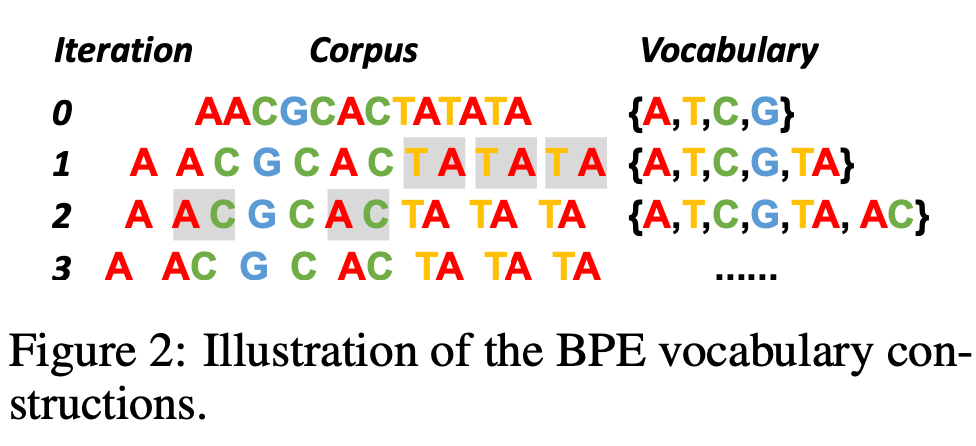
Byte-pair encoding process for DNA sequences
Strengths
- ✅ Adaptive Vocabulary - Learns optimal subunits from data
- ✅ Efficiency - Shorter sequences, lower computational cost
- ✅ Pattern Discovery - Automatically identifies frequent motifs
- ✅ Robustness - Handles sequence variations effectively
- ✅ Scalability - Works well with large datasets
Weaknesses
- ❌ Preprocessing Intensive - Requires corpus analysis for optimal merges
- ❌ Rare Pattern Loss - May miss infrequent but important motifs
- ❌ Domain Dependency - Vocabulary tied to training corpus
- ❌ Interpretability - Less intuitive than fixed k-mers
Implementation Example
from collections import Counter
import re
class DNA_BPE:
def __init__(self, vocab_size=1000):
self.vocab_size = vocab_size
self.merges = {}
self.vocab = set()
def get_pairs(self, word):
"""Get all adjacent pairs in a word"""
pairs = set()
prev_char = word[0]
for char in word[1:]:
pairs.add((prev_char, char))
prev_char = char
return pairs
def train(self, sequences):
"""Train BPE on DNA sequences"""
# Initialize with character-level tokens
vocab = Counter()
for seq in sequences:
vocab.update(list(seq))
# Iteratively merge most frequent pairs
for i in range(self.vocab_size - len(vocab)):
pairs = Counter()
for seq in sequences:
pairs.update(self.get_pairs(seq))
if not pairs:
break
best_pair = pairs.most_common(1)[0][0]
self.merges[best_pair] = f"{best_pair[0]}{best_pair[1]}"
# Update sequences with new merge
sequences = [seq.replace(f"{best_pair[0]} {best_pair[1]}",
self.merges[best_pair]) for seq in sequences]
self.vocab = set(vocab.keys()) | set(self.merges.values())4. Embedding-Based Methods
Word2Vec Embeddings
Overview: Treats k-mers as “words” and learns dense vector representations that capture contextual relationships between sequence elements.
Strengths
- ✅ Semantic Relationships - Captures biological similarities
- ✅ Dimensionality Reduction - Dense representations
- ✅ Transfer Learning - Pre-trained embeddings available
- ✅ Contextual Information - Considers k-mer neighborhoods
Weaknesses
- ❌ Large Dataset Requirement - Needs substantial training data
- ❌ Rare Pattern Issues - Poor performance on infrequent k-mers
- ❌ Fixed Context - Limited context window size
GloVe Embeddings
Overview: Analyzes global co-occurrence statistics of k-mers, capturing both local and global sequence relationships.
Strengths
- ✅ Global Context - Considers entire corpus statistics
- ✅ Stable Training - More consistent than Word2Vec
- ✅ Interpretable Relationships - Clear similarity metrics
Weaknesses
- ❌ Computational Cost - Expensive co-occurrence matrix construction
- ❌ Memory Requirements - Large matrices for big vocabularies
FastText Embeddings
Overview: Extension of Word2Vec that represents k-mers as bags of character n-grams, enabling understanding of subword information.
Strengths
- ✅ Subword Information - Captures sub-k-mer patterns
- ✅ OOV Handling - Manages unseen k-mers
- ✅ Morphological Awareness - Understands k-mer composition
Weaknesses
- ❌ Complexity - Higher computational overhead
- ❌ Parameter Tuning - Requires n-gram length optimization
📊 Specialized Encoding Approaches
5. Frequency-Based Encoding
Overview
Encodes sequences based on k-mer frequency counts, creating fixed-length vectors representing sequence composition.
Strengths
- ✅ Fixed Length - Consistent output dimensions
- ✅ Compositional Analysis - Captures sequence characteristics
- ✅ Simple Implementation - Easy to understand and implement
- ✅ Memory Efficient - Compact representation
Weaknesses
- ❌ Position Loss - No spatial information preserved
- ❌ Order Independence - Different sequences may have identical encodings
- ❌ Context Loss - No sequential dependencies
Implementation Example
from collections import Counter
def frequency_encode_dna(sequence, k=3):
"""Encode DNA sequence based on k-mer frequencies"""
# Generate all possible k-mers
nucleotides = ['A', 'T', 'C', 'G']
all_kmers = [''.join(p) for p in itertools.product(nucleotides, repeat=k)]
# Count k-mers in sequence
kmers = generate_kmers(sequence, k=k)
kmer_counts = Counter(kmers)
# Create frequency vector
freq_vector = [kmer_counts.get(kmer, 0) for kmer in all_kmers]
return np.array(freq_vector)6. Physicochemical Property Encoding
Overview
Incorporates biochemical properties of nucleotides (hydrophobicity, molecular weight, hydrogen bonding) into the encoding process.
Strengths
- ✅ Biological Context - Includes chemical properties
- ✅ Enhanced Prediction - Better for structural/functional tasks
- ✅ Multi-dimensional - Rich feature representation
- ✅ Interpretable - Clear biological meaning
Weaknesses
- ❌ Data Requirements - Needs comprehensive property databases
- ❌ Complexity - May not improve all tasks
- ❌ Domain Knowledge - Requires biochemistry expertise
📈 Comparative Analysis and Selection Guide
Performance Comparison Table
| Method | Sequence Length | Memory Usage | Training Time | Biological Context | Best Use Case |
|---|---|---|---|---|---|
| One-Hot | Short (< 1kb) | Very High | Low | None | Exact position analysis |
| k-mer | Medium (1-10kb) | Medium | Low | Local patterns | Motif discovery |
| BPE | Long (> 10kb) | Low | High | Adaptive patterns | Large-scale genomics |
| Word2Vec | Any | Low | High | Semantic | Functional prediction |
| Frequency | Any | Very Low | Very Low | Compositional | Sequence classification |
| Physicochemical | Short-Medium | Medium | Medium | Chemical properties | Structural prediction |
Selection Decision Tree
📋 Choosing the Right Encoding Method:
1. **Sequence Length**
- Short (< 1kb): One-Hot Encoding
- Medium (1-10kb): k-mer Tokenization
- Long (> 10kb): BPE or Embeddings
2. **Task Type**
- Position-specific: One-Hot Encoding
- Pattern recognition: k-mer or BPE
- Functional prediction: Embeddings
- Classification: Frequency-based
3. **Computational Resources**
- Limited memory: Frequency or BPE
- Limited time: One-Hot or k-mer
- High resources: Embeddings or Physicochemical
4. **Interpretability Requirements**
- High: One-Hot or k-mer
- Medium: Frequency or Physicochemical
- Low: Embeddings or BPE🔬 Practical Implementation Guidelines
Code Example: Complete Encoding Pipeline
import numpy as np
from sklearn.preprocessing import StandardScaler
from sklearn.model_selection import train_test_split
import pandas as pd
class DNAEncodingPipeline:
def __init__(self, method='kmer', **kwargs):
self.method = method
self.kwargs = kwargs
self.encoder = None
self.scaler = StandardScaler()
def fit_transform(self, sequences, labels=None):
"""Fit encoder and transform sequences"""
if self.method == 'onehot':
encoded = self._one_hot_encode(sequences)
elif self.method == 'kmer':
encoded = self._kmer_encode(sequences)
elif self.method == 'frequency':
encoded = self._frequency_encode(sequences)
else:
raise ValueError(f"Unknown method: {self.method}")
# Scale features
encoded_scaled = self.scaler.fit_transform(encoded)
return encoded_scaled
def transform(self, sequences):
"""Transform new sequences using fitted encoder"""
# Implementation depends on method
pass
def _one_hot_encode(self, sequences):
# Implementation here
pass
def _kmer_encode(self, sequences):
# Implementation here
pass
def _frequency_encode(self, sequences):
# Implementation here
pass
# Usage example
sequences = ["ATCGATCG", "GCTAGCTA", "TTAACCGG"]
labels = [0, 1, 0]
pipeline = DNAEncodingPipeline(method='kmer', k=3)
X_encoded = pipeline.fit_transform(sequences)
X_train, X_test, y_train, y_test = train_test_split(X_encoded, labels, test_size=0.2)🚀 Advanced Considerations and Future Directions
Hybrid Approaches
- Multi-scale encoding: Combining different k-values
- Ensemble methods: Using multiple encoding strategies
- Hierarchical representations: Incorporating sequence structure
Emerging Techniques
- Transformer-based encodings: BERT-like models for genomics
- Graph representations: Modeling sequence relationships as graphs
- Attention mechanisms: Learning important sequence positions
Performance Optimization
- Memory management: Efficient storage for large datasets
- Parallel processing: Scaling encoding for genomic databases
- GPU acceleration: Leveraging hardware for speed
🎯 Conclusions and Recommendations
Key Takeaways
- No Universal Best Method - Optimal encoding depends on specific task, data, and constraints
- Trade-offs are Inevitable - Balance between information retention, computational efficiency, and interpretability
- Preprocessing Matters - Quality of encoding significantly impacts downstream performance
- Domain Knowledge Helps - Understanding biology improves encoding choices
Practical Recommendations
- Start Simple: Begin with k-mer tokenization (k=4 or k=5)
- Validate Thoroughly: Test multiple methods on your specific dataset
- Consider Computational Constraints: Match method to available resources
- Preserve Interpretability: Choose methods that allow biological insight
- Monitor Performance: Track both accuracy and computational metrics
Future Research Directions
- Attention-based models for learning optimal encoding strategies
- Multi-modal approaches integrating sequence and structural data
- Transfer learning from pre-trained genomic models
- Automated encoding selection using meta-learning approaches
📚 References and Further Reading
Zhou, Z. et al. (2023). DNABERT-2: Efficient Foundation Model and Benchmark for Multi-Species Genome. arXiv preprint arXiv:2306.15006.
Sennrich, R. et al. (2016). Neural Machine Translation of Rare Words with Subword Units. ACL 2016.
Mikolov, T. et al. (2013). Efficient Estimation of Word Representations in Vector Space. arXiv preprint arXiv:1301.3781.
Pennington, J. et al. (2014). GloVe: Global Vectors for Word Representation. EMNLP 2014.
Bojanowski, P. et al. (2017). Enriching Word Vectors with Subword Information. TACL 2017.
This comprehensive guide provides both theoretical understanding and practical implementation details for DNA sequence encoding. The choice of encoding method is crucial for genomic machine learning success - choose wisely based on your specific requirements and constraints.
For more advanced genomics and AI content, explore our AI for Genomics and Machine Learning sections.
Tags: #Bioinformatics #MachineLearning #DNASequencing #ComputationalBiology #Genomics #DataScience #SequenceAnalysis #AIforGenomics